初探單細胞定序

Quick look
單細胞定序已經在近年成為各大實驗室不可或缺的關鍵技術之一,讓我們從過去將組織混雜在一起的bulk RNA-seq精細化到單細胞的解析度,也讓我們可以針對一顆腫瘤做細部的分類,、甚至推測細胞分化的路徑,今天我們要讀的文章算是比較舊的nature protocol review article (2018),但針對我們想要做單細胞定序分析的人來說是很推薦的入門讀物之一。
scRNA-seq 席捲科學界
scRNA-seq 顯著增加了我們對組織、器官和細胞之間複雜交互作用的了解。隨著自動化處理流程的演進與微流體(microfluidic)技術的發明,scRNA-seq的延展性(scalability)才能大幅的提升。
過往的實驗設計通常是基於某一個假說或是假設(hypothesis),但若是這個假設錯誤或是不成立,就要花額外的時間從另外一個假設著手,這種hypothesis-driven的方式比較費時。自從有了scRNA-seq後,我們可以一探每一顆細胞的全基因組、蛋白質組甚至是表觀遺傳基因組,從hypothesis-driven過渡到data-driven的實驗設計,大幅降低bulk情形下帶來的biased analysis。
但因為scRNA-seq的研究太專一化,不同的樣本有截然不同的處理步驟,而數據分析也依據不同的需求而有所不同,因此在研究設計上無法將一套準則套用在所有的實驗上,但也因為這個限制,近年有層出不窮的protocols、tools或是網頁工具出現,試圖來優化及改善整條從樣本製備到數據分析的過程 (圖一)。
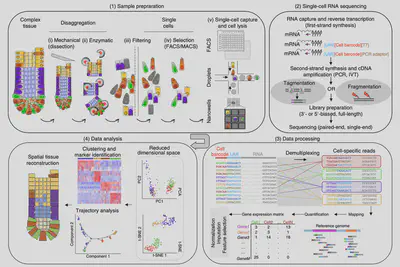
樣本製備
樣本本身的品質對整個scRNA-seq的成敗是最關鍵的。在過去,大部分都要求使用新鮮的活細胞,但實務上如果要在取出每個腫瘤後立即分解成單細胞懸浮液其實頗具挑戰性,因此目前有不少protocol是允許使用固定後的腫瘤或細胞,也允許冷凍後仍完整的細胞核RNA,一來可以讓我們可以好好計畫樣本製備的流程,二來也不影響後續數據的分析,可以參考目前10X Genomics釋出的GEM-X FLEX protocol。
其他一些注意事項如下:
- 使用
無核酸酶試劑和耗材。 - 減少樣本溶液的
轉移與離心操作以避免細胞損傷。 - 篩選較大的細胞團塊和細胞死亡後的雜質與碎片。最好在
30分鐘內用酵素分解細胞團塊,避免團塊聚集。 - 適合的懸浮液緩衝液組成:
無鈣、鎂的PBS。含牛血清白蛋白以減少聚集。比較敏感的細胞、幹細胞可能需要其他的緩衝液來增加存活。
細胞懸浮液的製備
血液樣本可以用密度梯度離心(density centrifugation)的方式來分離,例如Ficoll-Paque 或 Histopaque-1077 的方式來捕捉特定單細胞,但是實體組織必須要利用機械(mechanical)或是酵素來分解組織塊來取得單細胞懸浮液。
機械方式:可以用剪刀或是剃刀將組織切成小碎塊,通常約大小 1mm x 1mm x 1mm,才能增加與酵素接觸的表面積。酵素分解:切成小塊後,要接著用酵素做分解,針對不同的組織有不同的酵素組合,可以參考圖二。
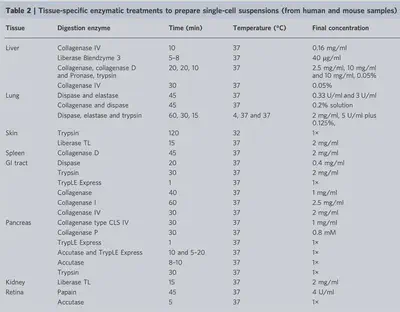
值得注意的是,在樣本製備的過程中,活的細胞有可能會因為過程中遭遇的stress而導致某些反應性基因的表現改變,因此過程中需要盡可能地減少stress。
另外就是針對像神經元所在的組織中,神經元彼此之間交聯的程度有可能會導致細胞分離的過程不完全。針對這個問題可以考慮破壞細胞膜的方式來取得完整的細胞核做分析,而用細胞核內部的RNA做分析雖然會降低每顆細胞最終的解析度,但是已能提供足夠訊息來解析細胞態 (cell type deconvolution)。
單細胞捕獲
做單細胞定序前,當然需要想辦法捕捉到單顆細胞,而目前有許多不同的方式:
- Microdissection
- Pipetting
- Fluorescence-activated cell sorting (FACS)
- Microfluidics
後面兩個技術為high-throughput,可以有效率的捕獲大量的單細胞。FACS 可以將帶有特定螢光的細胞挑出來,並收集到微孔板(microtiter plate)中;而microfluidics 是利用integrated fluidic circuits (IFC)、油滴或是奈米板(nanowell)、來同時收集及處理細胞,減少試劑的使用。有些時候為了降低背景噪音及優化定序的表現,可以在使用microfluidic 系統前先用FACS或MACS(magnetic-activated cell sorting)來移除死細胞或是雜質。
樣本大小與組成
雖然取得unbiased 細胞組成(盡可能捕捉到全部)很重要,但有時候太大或太小的細胞反而會卡在微流體系統或是被FACS忽略,因此有時候研究反而會針對某些特定的細胞群體,如免疫細胞去做富集,或是去掉血球細胞(CD45+)。
FACS的技術可以藉由螢光強度或細胞大小(FACS可以提供的訊息)來對細胞做編列(indexing),讓我們可以知道細胞在sorting時的位置與螢光強度,讓後續做transcriptomic profiling 時可以追蹤細胞標記。
除了FACS系統,CITE-seq(圖三)利用oligonucleotide標記的抗體辨識細胞表面的記號(epitope),這個抗體專一性oligonucleotide sequence 接上了一條poly(A) tail 及一段特殊條碼,讓我們在scRNA-seq library construction時可以追蹤epitope。
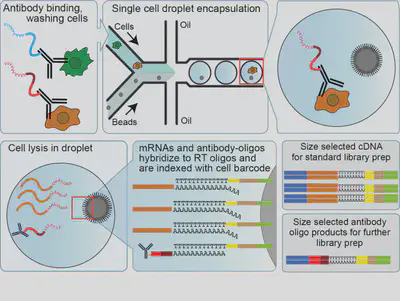
細胞數量
另外實驗的細胞數量需要多少也是很重要的參數,須要考慮樣本的異質性與感興趣細胞的出現頻率,一般來說,有兩種情況需要較多的細胞:
樣本異質性高: 所以需要較多的細胞數量來解構所有的次群體。感興趣的細胞很罕見: 所以需要較多的細胞數量來增加他出現的次數。
針對同質性高的樣本並不是說細胞數量可以減到很少,因為足夠的細胞數量才能增加統計效力。
目前也有工具可以幫我們估計我們需要多少細胞數量,如Satija 教授開發的網路工具howmanycell。
針對目前GEM-X FLEX 提供的protocol,因為是基於探針來偵測基因的表現,所以最多只能提供50萬個細胞的探針,更多的細胞會被浪費掉,所以在實際操作的時候需要詳細閱讀protocol。
樣本保存
過去大部分scRNA-seq的步驟都要求取得新鮮的活細胞來做定序,但是取得腫瘤後立即處理樣本其實頗具挑戰性,也很吃人力和時間成本,如果又沒有FACS的專門的儀器,會變得相當尷尬。
此外,雖然可以在不同天處理腫瘤,但是我們都知道這樣會引入Batch effect,因此同一天處理樣本是最理想的狀況,想當然底下的研究助理會想哭吧…。
有鑑於此,不少的冷凍保存(cryopreservation)技術也開發出來,讓我們可以使用冷凍後的樣本做處理。而研究顯示,在-80度或是液態氮保存的樣本在解凍後,仍然保有完整的RNA與基因表現,但目前仍不建議對樣本進行反覆的冷凍與解凍。
而針對急速冷凍(snap-freeze)的樣本,因為冰晶(crystal)會破壞細胞膜,多數細胞並不完整,只能利用nuclei 來取得scRNA-seq。
單細胞定序
單細胞定序主要有四個步驟:
RNA分子的捕獲(capture)放大(transcriptome amplification)建立定序庫(sequencing library)定序(sequencing)
RNA molecule capture, reverse transcription and transcriptomics amplification for sequencing library construction
大多的單細胞的RNA分子捕獲利用poly(A)-tailed RNA來辨識mRNA,針對total RNA 也有其他特殊的方式,但比較少用。
所以我們只要設計一段poly(T) oligonucleotide就可以收集所有mRNA,但是必然會忽略掉數量也很多的rRNA與tRNA,在捕獲之後,RNA片段會被反轉綠成cDNA,接續進行放大與定序庫的建立。
在設計oligonucleotide的時候,會插入專一的單細胞編碼(single cell barcode),讓後續在定序時,可以做pooling與multiplex。
除了cell barcode外,還會插入約12的核苷酸的unique molecular identifier (UMI),用來去除後續cDNA擴增後帶來的擴增噪音,是單細胞分析裡面很關鍵的技術。關於UMI如何作用,可以參考這個網站。
而針對cDNA的擴增,基本上有兩種方式:
- PCR:所需步驟較少,但是對RNA的定量會引入較多的擴增偏差。
In vitro transcription (IVT):利用線性擴增,因此引入的擴增偏差較少,但需要的後續步驟較多。
圖四為各種平台的技術概況: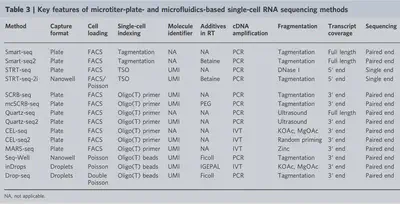
Full length vs 3’ or 5’ transcript sequencing
轉錄的過程可以分為全長(full length)或是針對5’/3’端來做定量(digital counting),這兩個方式的差別在於:
Full length: mRNA的整段都可以解碼出來,因此適合用於須要了解splice variant或是alternative splicing的研究。T與B細胞的受體genotype 也可以藉由full length sequencing 來還原。 然而這種技術因為並沒有在處理初期插入UMI,所以並不能做indexing,因此實驗成本會較高。3'/5' sequencing: 因為PCR先天的限制,在mRNA的尾端無法完整進行下去,因此會喪失掉尾端的訊息,但如果只在乎基因的表現量,這樣的限制並不會影響實驗的目的。此外,因為在處理的過程中可以加入cell barcodes,所以有利於indexing與multiplex,減少成本。
scRNA-seq methodology
這邊僅針對目前最常用的微流體系統來做描述,想了解其他方法,如microtiter plate-based 或split-pool barcoding-based的可以另外搜尋文獻。
Microfluidic system-based approaches
微流體系統的優勢在於可以高通量的針對單細胞做處理,具有很高的技術延展性(scalability),彌補microtiter-based 方法的不足,同時增加了cDNA的產量。
最初用來做scRNA-seq的微流體系統使用拋棄式微流體晶片,完成單細胞裂解、核酸純化、反轉錄作用和擴增放大流程,最後將單細胞之終產物存放至獨立槽中,進行後續分析,Fluidigm C1就是使用這種方式。但早期的晶片只有96個細胞捕獲位點,後來新的技術(C1 HT-IFC)才增加到了800個位點,並允許早期編碼(early-indexing),讓細胞可以混合在一起做後續分析,大幅降低了成本。
為了進一步增加捕獲的細胞數量,微流體技術發展為更具擴展性的開放式奈米孔系統。例如,STRT-seq-2i運用具有9600個位點的奈米孔平台,通過限制性稀釋或特殊的FACS技術排序並加載細胞。此外,奈米孔陣列的形式允許我們透過影像來觀察一個孔裡面是否有出現兩個以上的細胞(doublet)。
另一個奈米孔技術為Seq-Well,可在捕獲高達86000個細胞並進行反應,原理是將帶有條碼的珠子先裝到奈米孔中,然後細胞通過限制性稀釋進入捕獲位點。隨後陣列被密封以進行細胞裂解和RNA的捕獲,最後將固定的RNA分子集中製備3’端library。然而,儘管這種技術可以通過顯微鏡監測細胞,但帶有條碼的珠子因為隨機分佈的特性使得影像的整合困難,此外也需要經驗豐富的使用者以保證結果的重現性和品質。
雖然IFC和奈米孔方法在高通量方面具有擴展性,但它們的反應位點數量還是很有限。液滴系統通過將細胞封裝在液滴中克服了這一限制。此方法可以捕獲的細胞數量隨著使用的乳液(emulsion)體積的上升而上升,並且能高速生成大量液滴,適用於大規模scRNA-seq實驗。此外,可以調整液滴大小以減少細胞捕獲過程中的引入的潛在偏差。由於條碼隨也是隨機導入液滴中,因此該方法也無法透過影像來偵測捕獲的即時狀況。
關於液滴技術目前有兩種:
inDrops利用水凝膠珠(hydrogel bead),上面帶有poly(T)引子與特殊細胞編碼,在捕獲細胞後,通過光來釋放引子提高分子捕獲效率並啟動液滴內的反轉錄反應,這項技術可以有大於75%的細胞捕獲率,因此很適合細胞數量較少的樣本。目前使用這種技術的廠商有:1CellBio。Drop-seq方法則使用帶有隨機條碼的珠子,在細胞裂解和RNA捕獲後,液滴破裂並且合成cDNA,接著進行3’端文庫製備。相比inDrops,Drop-seq因採用雙重限制稀釋,細胞捕獲效率較低,目前採用Drop-seq系統的為Dolomite Bio與Illumina(ddSEQ)。
我們來看一下一個比較表(圖五)
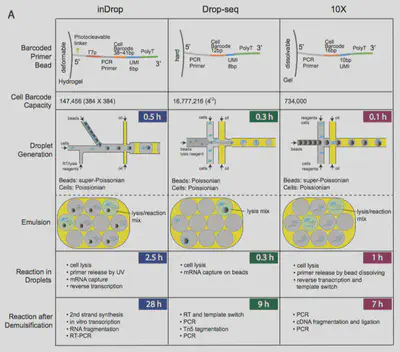
不難發現10x Genomics系統比較類似Drop-seq,但10X Genomics使用可溶解的beads,而可溶解beads的的優點:
- 當beads溶解後,引子均勻地釋放到液滴中,提供了一個更加均一的反應環境。這能夠提高條形碼與mRNA分子的結合效率,從而提升cDNA合成的成功率
- 條碼在液滴中釋放後,分佈更均勻,減少了因beads表面接觸限制而導致的偏差,使得mRNA條形碼標記的均勻性更高。
- 利用同步分配技術(例如微流體設備中的精準控制),可實現更高的單細胞分離效率,降低雙細胞(doublet)的比例。
- 可溶解beads設計可減少環境RNA的污染,因為在釋放條形碼後,RNA分子更快地被捕獲並固定。
Library 的準備與定序
在短序列片段(short-read sequencing)的應用中,擴增後的cDNA(經由PCR)或RNA(經由IVT)會在加入sequencing adaptor 之前先進行片段化。片段化可透過酵素(如tagmentase或DNase)、化學方法(如使用鋅、醋酸鉀或醋酸鎂)或機械(如超聲波)來達成。接著,基於3’或5’的文庫會使用針對轉錄起點或終點的引子進行擴增。在此步驟中,可以加入一個pool-specific index,所以可以進行multiplexing。
Full-length的方法僅在片段化之後才引入細胞專一的barcode,這會阻礙細胞在處理的早期階段進行混合。除了STRT-seq外,scRNA-seq文庫需要使用paired end sequencing,其中一端提供轉錄信息,另一個端則讀取single cell barcode與UMI。STRT-seq則將cell barcode與UMI嵌入到5'轉錄末端,因此序列之間並沒有被poly(T) oligonucleotides分隔開,這使得單一端點即可同時獲取細胞、分子與轉錄的相關信息。
10X Genomics 的UMI與cell barcode與olgo(dT)都在同一端(5’ end),捕獲mRNA後,transcript information往3’ end延伸,所以光sequence 5’ end 只能得到barcode與UMI訊息。
微流體的實驗因為捕獲的細胞數量較大(RNA分子捕獲率相對較低),通常進行較淺的定序深度(<100,000 reads/每細胞),而較高的定序深度(~500,000 reads/每細胞)則適用於microtiter plate。然而,雖然增加定序深度可以提高解析度(檢測更多基因及低表現的基因),我們通常不會將單細胞文庫定序到飽和的程度。Full-length轉錄組的splice variant需要更深度的定序,才能更準確地解析轉錄體的變異。
其他技術考量
Cell doublets
大多數基於微流體的方法有一個潛在的問題:每個反應位點(奈米孔或液滴)可能同時捕捉到兩個細胞,這導致它們擁有相同的barcode,這種現象稱為doublet,而doublet rate可以透過物種混合實驗(species-mixture)來估算。
Doublet的產生源於細胞在反應位點中的隨機分佈,其發生率與細胞懸浮液的濃度有關。對於Chromium系統,在每個液滴通道最多建議的10,000個細胞裝載量下,doublet rate呈線性關係(符合Poisson分佈),其推算的doublet rate範圍為2%(2,500個細胞)至8%(10,000個細胞)。其他微流體方法的doublet rate也類似:Drop-seq的doublet rate為0.36–11.3%(每微升12.5–100個細胞),InDrops為4%,Seq-Well則為1.6%。
在較高稀釋條件下,doublet rate會降低,但這也表示每個實驗捕捉的總細胞數減少,進而提高每個細胞的試劑成本。研究人員可以部分克服這個問題,方法是同時捕捉來自不同個體的樣本,透過基因型差異來區分來源,從而識別doublet個別的來源。具體而言,可以利用RNA定序reads中的單核苷酸多樣性(SNP)來判斷細胞的來源,並區分在同一個batch中處理的樣本。然而,這種流程僅在實驗設計中涉及不同人類個體或具有明顯遺傳背景差異的時候才可行。
目前,尚無能夠可靠辨識doublet的計算方法,因此必須透過實驗設計來將doublet rate降至最低。Doublet可能對數據解釋產生顯著影響,因為混合在一起的轉錄組可能被誤解為中間的細胞狀態,影響判讀。
Cell capture efficiency
細胞捕獲效率是實驗中一個重要的考量因素,特別是在涉及primary cell或rare samples的實驗中。而獲得條碼的細胞數量與進入下游分析的樣本比例直接相關。
FACS方法的捕獲效率受限於設備在不同孔之間移動所需的時間。為了最大化FACS方法的捕獲率,可以稀釋並以低速率(例如每秒100個細胞)對細胞懸浮液進行sorting。
微流體技術在捕獲效率上與FACS有明顯的差異,主要取決於細胞和珠子的裝載機制。HT-IFC系統最多能從6,000個注入的細胞中捕獲800個細胞。
而在奈米孔系統中,細胞通過極限稀釋法進行裝載(無需分選),細胞靠重力進入反應位點,通常具有較高的效率。例如,在Seq-Well系統中,將10,000個細胞加入到陣列表面後,約有3,000個細胞能被捕獲。
對於液滴式系統,細胞進入分析的速率與珠子的裝載效率直接相關。當大多數液滴中含有條碼珠時,細胞捕獲效率最佳(例如inDrops系統)。相反,如果珠子和細胞通過極限稀釋進行封裝,大部分細胞無法進入含有珠子的液滴中,導致較低的捕獲效率(例如Drop-seq系統)。
Cost
scRNA-seq實驗的總成本主要由三個部分決定:設備、試劑和測序。對於大多數方法而言,scRNA-seq文庫製備的成本隨著細胞數量呈線性增加;然而,自製液滴法是個例外。各種方法和不同研究機構之間,每個細胞的實際成本差異很大。微流體系統的成本通常較低(每個細胞低於0.30美元),而early indexing plate-based的3’端digital counting的方式成本較高(約每個細胞1–2美元)。而late indexing plate-based的full-length轉錄組分析成本更高,即便反應體積較小,每個細胞仍需約8–12美元。
然而,透過使用非商業化的tagmentase或最小化反應體積,並針對plate-based方法採用自動化工作流程,可以有效降低成本。此外,微孔板可進行運輸和儲存,這使得樣本採集地點可以與scRNA-seq流程分離,從而將昂貴的設備集中於核心實驗室,實現最佳的資源管理。
儘管文庫製備的成本正在迅速下降,測序成本卻逐漸成為主要因素。具有更高分子捕獲效率的方法能夠產生更複雜的測序文庫,因此即便在較低的測序深度下也能提供豐富的資訊。因此,更高效的scRNA-seq方法雖然增加了文庫製備的成本,但可以透過降低整體測序成本來進行彌補。
數據處理
資料處理包括將原始測序轉換為基因表達矩陣的所有步驟,這些工作流程與傳統bulk RNA-seq的處理流程類似。當生成FASTQ檔案並使用工具如FastQC進行quality control後,下一個重要步驟是使用細胞條碼對reads進行去重複索引(de-multiplexing)。
去重複索引後的reads會使用比對工具(如TopHat和STAR)與參考基因組進行比對,其中STAR準確性優異,對splice variant的辨識也相當靈敏。近年來的新型比對工具經過優化,能在不損失準確性的前提下,快速處理大規模數據集。例如,Kallisto透過pseudo-alignment,而非逐個鹼基的比對,大幅縮短了比對時間,快上兩個量級。
在最終的處理步驟中,經比對的reads在定量後會轉成基因表達矩陣。
RSEM、Cufflinks 和 HTSeq 適用於full-length轉錄組數據的定量。
而對於UMI標記數據類型,如考慮UMI序列中出現的測序錯誤,則可以使用UMI-tools。
除了專門處理各個步驟的工具之外,還有整合的單細胞數據處理工作流程,這些流程結合了比對和定量步驟,並包含對reads和細胞的quality control。例如,Ilicic 等人開發的一個支援多種比對和定量工具,並包括過濾低質量細胞的模組。
Scater 提供了一個的組織化的工作流程,將原始測序reads轉換為單細胞表達集合(SCESet)類別 (class),這是一種數據結構,有助於數據的處理和分析。
此外,還有其他工作流程:
zUMI、scPIPE和SEQC適用於UMI數據,這些流程是基於特定protocol設計的。Cell Ranger則是專為Chromium系統設計的流程。- 最後,scRNA-tools資料庫提供了一個完整的清單,列出用於數據處理和分析的計算工具。這些方法根據
分析任務進行分類,研究人員可以根據所需的分析類型選擇合適的工具。
Normalization
scRNA-seq數據存在不少噪音與變異性,來源包括技術處理流程、如dropout events、biased amplification與不完整的文庫建立。技術性變異也包含不同的樣本處理時機、時間點與儀器等因素。除此之外,生物因素也佔了不小因素,導致分析上的複雜度增加,如不同的細胞大小、RNA內容物、不同的細胞週期與性別差異等。因此,數據的標準化是產生有意義數據前最重要的一環。
過去真對bulk RNA-seq開發的標準化方法,如log-expression、trimmed mean M-values與upper-quartiles可以拿來用在scRNA-seq的分析上,但近期也針對這種特殊的數據出現的不少的標準化方法,如SCnorm與SCRAN,可以針對組間與細胞因素做標準化。
然而,對大規模的數據變異來源,直接建立模型是比較標準的做法,例如將干擾因素(confounding factor)當作變數納入回歸模型,來校正數據的分佈。
例外對batch effect來說,通常是用視覺化的方式如PCA來偵測,而kBET利用k nearest neighbors,量化組內與組間的batch effect,發現結合(log normalization或SCRAN pooling)與(ComBat或是limma)等可以提供最佳的校正後數據,同時保存生物學意義。
如果數據收集時間的不同、不同個體、或是來自不同的scRNA-seq的技術都會導致數據出現嚴重的batch effect。Haghverdi等人提出基於mutual nearest neighbors的方式,利用共同的subset population來校正不同實驗間的batch effect。此外,Biscuit (Bayesian inference for single-cell clustering and imputing)利用基因表現相似性與共表達特徵來推論細胞群集,藉此校正細胞間的技術性變異。
常用來處理單細胞數據的Seurat也有針對常見變異來源做校正與整合,也推出新的方法來找出不同數據集共同的細胞族群及進行 比較性分析。
Imputation and gene selection
因為scRNA-seq的數據集非常稀疏 (sparse),會影響細胞表型的辨別與判讀,例如很多沒有表現的基因或是dropout events會導致數據矩陣出現很多0。為了降低矩陣的稀疏性,缺失的transcripts可以用估計的方式來補足,例如MAGIC使用diffusion map 來還原缺失的數據。而scImpute利用擬合一個混合模型來學習基因的dropout 機率,並使用相似的細胞來推論可能的dropout events。
針對樣本的異質性,常見的方式是分析不同數據集之間高表達變異度的基因,先完整的用特徵選取來移除噪音較多的基因不僅可以增加訊號與雜訊的比例,也可以降低計算的複雜度。常用的策略是探討mean transcript abundance與數據的離散程度 (dispersion)、負二項分佈 (negative binomial distribution)的離散參數 (dispersion parameter) 或是總變異 (total variability) 比例之間的關係。
高變異度的基因,在去除技術性外在因素之後,可以用來解釋生物性上的差異與生物異質性。
Data analysis
scRNA-seq實驗的許多目的是評估樣本的異質性及找出新的細胞類型與狀態。我們通常可以利用共表達矩陣與細胞相似性的clustering來達成。而後續細胞的聚集可以用空間降維的方式如PCA或t-SNE來投影與解讀,另一個近年常用的UMAP克服了前面兩種方法的限制,保存了整體的數據結構與細胞的pseudo-temporal ordering,而且速度更快。即使這些降維方式可以輔助初期的數據判讀,但是需要更強大的聚類演算法來定義細胞的次群體(subpopulation)。
雖然監督式演算法(如Monocle2)可以利用先驗假設和細胞的典型標記來聚類,但在大多數情況下,若能使用無假設(hypothesis-free)的非監督式聚類通常是更好的選擇。一種常用的非監督式演算法是階層聚類(hierarchical clustering),該方法在沒有預設聚類數量的情況下提供一致的結果。階層聚類可以採用聚合式(bottom-up)或分裂式(top-down)的方式進行,分別通過連續的合併或分裂群集來完成。層次聚類的工具有 PAGODA、SINCERA 和 bigSCale。此外。
另一種適合的非監督式聚類算法是 k-means,其基本原理如下:
設定k個聚類中心(centroids),將每個細胞分配到最接近的聚類中心,根據每個聚類群內的細胞均值重新計算聚類中心。
不斷重複這些步驟,直到聚類穩定。例如,SC3 工具結合了 k-means 和層次聚類,能夠為細胞提供準確且穩健的聚類結果。
其他非監督式聚類方法(如 SNN-Cliq 和 Seurat)則使用基於圖的聚類(graph-based clustering)。該方法通過構建圖形,將細胞作為節點,並根據表達相似性建立邊,然後將圖形分割成相互連接的準團塊(quasi-cliques)或社群(communities)。
聚類可以直接基於基因表達值或更為處理過的數據類型(如主成分或相似性矩陣)來進行。其中,相似性矩陣在聚類分離上具有更好的效果。
而群集穩定性可以通過以下方式來測量:
- 重抽樣方法(如 bootstrapping)。
- 基於群集內細胞相似性(如 silhouette index)。
而為了支持群集結果的再現性(replicability),可以使用調整後的 Rand 指數(adjusted Rand index)來比較不同的演算法。最後,可以將群集使用降維(DR)演算法(如 PCA、t-SNE)投影到二維或三維空間中,並通過顏色編碼來表示不同的群集。
可以通過對群集進行差異基因表達分析(differential gene expression analysis)來識別能夠區分細胞亞群的標誌基因(marker genes)。例如,可以使用模型的方法,如 SCDE、MAST 和 scDD,這些方法通過混合模型處理數據的雙模性(bimodality)。針對個別基因,可以評估它們是否能作為細胞身份的二元分類器(binary classifiers)。常用的方法包括基於零膨脹數據(zero-inflated data)的 ROC(Receiver Operating Characteristic) 或 LRT(Likelihood Ratio Test) 測試。
另一個scRNA-seq的重要應用是軌跡推斷(trajectory inference),該方法通過預測的分化路徑(pseudotime)對細胞進行排序,以估計動態過程。常用的算法包括反向圖嵌入(reversed graph embedding,例如 Monocle2)和最小生成樹(minimum spanning tree,例如 TSCAN)。為進一步促進結果的解釋性,可以使用工具如SCENIC,該工具可以讓我們了解胞亞群中的活性調控網絡。這種分析能幫助識別活躍的轉錄因子,最終找出造成細胞異質性的細胞機制。
對於群集註解(cluster annotation),工具如scmap 可以通過將一個數據集中的細胞投影到另一個 scRNA-seq 實驗的細胞類型或個別細胞上,來實現跨實驗的數據比較。而使用如 bigSCale 這樣的細胞卷積工具,scRNA-seq 分析的規模可以擴展到數百萬細胞。
最後,單細胞數據可以通過實驗方法來呼應組織的空間分佈,或者通過細胞的擬空間排序(pseudo-spatial ordering)來進行分析和解釋。
為了使scRNA-seq數據公開讓大家使用,可使用專門的數據存儲和共享庫。Gene Expression Omnibus(GEO)是常用的數據存取平台,用於提供原始數據和經過進一步處理的數據格式,例如基因表達定量矩陣。一些大型項目(例如人類細胞圖譜(Human Cell Atlas))建立了專門的數據協調平台,進一步簡化了數據查詢和調用的過程。
未來方向
scRNA-seq正在快速發展。每次實驗可以分析的細胞數量已經增加到數十萬以上,顯著提升了罕見或是過度型態細胞的檢測能力和解析度。然而,高通量技術的代價是分子捕獲率降低,因此未來的方法需要在細胞數量和細胞解析度之間實現更好的平衡。
目前,組織和整體生物體級別的項目通常採用跳傘式(sky-dive)的實驗策略,意即先通過數千個細胞來建立低解析度的圖譜,估算樣本的異質性,然後利用高效的 scRNA-seq 方法對目標細胞類型進行深入分析,以獲得更高的單細胞解析度。未來,高解析度的圖譜將允許研究者在現有數據中進行放大,從而避免昂貴且耗時的樣本重處理。