單細胞RNA-seq數據分析教學-3:整合單細胞數據集
Quick look
在當前的研究實務中,很少只進行一次scRNA-seq實驗並僅產生一份scRNA-seq資料。這主要有兩個原因:首先,目前的scRNA-seq技術僅能在單次實驗中對有限的樣本提供訊息;若要跨實驗、多條件地測量多個樣本,通常需要將多次實驗所得的scRNA-seq資料進行聯合分析(Joint analysis)。儘管已有如cell hashing這樣的實驗策略,以及像demuxlet與scSplit這類可對樣本進行計算區分的方法 (是在同一次 scRNA-seq 實驗中混合多個樣本(例如不同病人的細胞)後,根據基因型或表現資料,將單細胞正確歸類到原始樣本身上),在製備與定序scRNA-seq文庫時可以將多個樣本混合,但某些步驟(如組織分離, Tissue dissociation)仍然不可避免地需要針對不同樣本個別進行。因此,就如同在處理bulk RNA-seq資料時一樣,批次效應(batch effect)通常是分析中必須解決的一個關鍵干擾因素。
在本教學的此部分,我們將介紹幾種scRNA-seq資料整合方法。我們將使用範例教學中的DS1資料集與DS2資料集進行展示。值得注意的是,目前尚未有一種在所有情境下皆最優的整合方法,因此實務上重要的是嘗試不同的方法並比較它們的效果,最終選擇最適合該特定資料集的方法。
載入資料
我們從匯入Seurat套件及儲存的Seurat物件開始:
library(Seurat)
library(dplyr)
library(patchwork)
seurat_DS1 <- readRDS("DS1/seurat_obj_all.rds")
seurat_DS2 <- readRDS("DS2/seurat_obj_all.rds")
步驟1:合併兩個資料集
首先,有可能批次效應並不大,因而無需進行整合。因此我們應先觀察兩個資料集的表現,方式是先將它們合併:
seurat <- merge(seurat_DS1, seurat_DS2) %>%
FindVariableFeatures(nfeatures = 3000) %>%
ScaleData() %>%
RunPCA(npcs = 50) %>% #需要先run PCA,然後用PCA的資訊喂給UMAP or TSNE
RunUMAP(dims = 1:20)
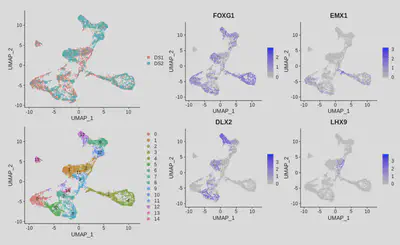
plot1 <- DimPlot(seurat, group.by="orig.ident") #用初始計畫名稱分類
plot2 <- FeaturePlot(seurat, c("FOXG1","EMX1","DLX2","LHX9"), ncol=2, pt.size=0.1)
plot1 + plot2 + plot_layout(widths = c(1.5, 2))
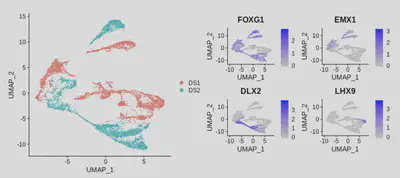
顯然,兩個資料集在UMAP嵌入圖上彼此分離。然而,marker基因的表現模式顯示,兩個資料集確實共用了相當多的細胞類型。理想情況下,相同細胞類型的細胞應該彼此混合。但由於批次效應,這並未發生,因此我們需要進行資料整合。整合的目標是讓相同類型的細胞能在兩個資料集中互相混合,而不同細胞類型或狀態的細胞則仍能區分。
- 這邊merge的數據集應該要是已normalize但還未scale的數據。
在這裡我們將嘗試以下幾種整合方法:
Seurat
Harmony
LIGER
MNN
步驟2-1:使用Seurat進行資料整合
Seurat 內建資料整合流程簡要來說,它首先對待整合的資料集使用 Canonical Correlation Analysis(CCA),將它們各自旋轉,以最大化兩者的共變化(canonical correlation)。換句話說,Seurat 使用 CCA 尋找能最大化資料集之間相似度的投影方式。接著,Seurat引入錨點(anchor)機制,尋找在兩個資料集中的細胞配對錨點。這些錨點是指在CCA空間中互為最近鄰的細胞對,且彼此在各自資料集中的鄰居傾向於也彼此鄰近。這些被錨定的細胞被視為跨資料集的對應細胞,接著會計算轉換矩陣,並用於調整其中一個資料集的表達值。
欲使用Seurat進行整合,首先必須對每個待整合資料集進行正規化並找出高變異基因(若尚未完成):
seurat_DS1 <- NormalizeData(seurat_DS1) %>% FindVariableFeatures(nfeatures = 3000)
seurat_DS2 <- NormalizeData(seurat_DS2) %>% FindVariableFeatures(nfeatures = 3000)
接著,尋找資料集間的anchors。在這一步,Seurat接受一個Seurat物件清單作為輸入。請注意,Seurat支援超過兩個樣本的整合,只需將它們放入list中:
seurat_objs <- list(DS1 = seurat_DS1, DS2 = seurat_DS2)
anchors <- FindIntegrationAnchors(object.list = seurat_objs, dims = 1:30)
- 註:dims參數決定了要使用多少個CCA元件,可視需要微調。
然後將這些anchor傳入IntegrateData函數以進行表達層次的修正:
seurat <- IntegrateData(anchors, dims = 1:30)
執行IntegrateData會產生一個新的Assay(預設名稱為integrated),其中儲存經批次修正的表達矩陣。原始(未修正)表達值並未消失,而是保留在原本的Assay(預設為RNA)中。整合完成後,Seurat物件的預設Assay會自動設為integrated,但也可透過以下方式切換:
DefaultAssay(seurat) <- "RNA"
接著,使用整合後的Seurat物件進行後續分析,與第一部分的處理流程相同,只需略過正規化與高變基因找尋這兩步:
seurat <- ScaleData(seurat)
seurat <- RunPCA(seurat, npcs = 50)
seurat <- RunUMAP(seurat, dims = 1:20)
seurat <- FindNeighbors(seurat, dims = 1:20) %>% FindClusters(resolution = 0.6)
saveRDS(seurat, file="integrated_seurat.rds")
需要特別注意的是,雖然tSNE/UMAP嵌入與分群應使用整合後的Assay(integrated),但這些經修正的表達值不再適合作為定量分析依據。因此,進行群集標記基因鑑定或視覺化時,建議切換回原始Assay(RNA):
DefaultAssay(seurat) <- "RNA"
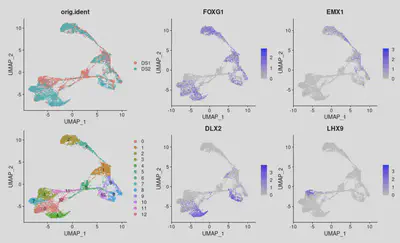
plot1 <- UMAPPlot(seurat, group.by="orig.ident")
plot2 <- UMAPPlot(seurat, label = T)
plot3 <- FeaturePlot(seurat, c("FOXG1","EMX1","DLX2","LHX9"), ncol=2, pt.size=0.1)
((plot1 / plot2) | plot3) + plot_layout(width = c(1,2))
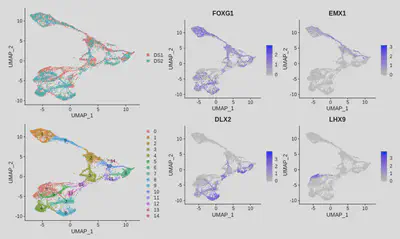
雖不完美,但這樣已能使兩個資料集更具可比性。
若想進一步優化結果,可調參數包括:
FindVariableFeatures中的nfeatures參數:影響整合用基因集的選擇。
FindIntegrationAnchors中的
anchor.features、dims、k.anchor、k.filter、k.score等。IntegrateData中的dims參數。
步驟2-2:使用Harmony進行資料整合
除了Seurat,現在已有更多可用的資料整合方法。Harmony就是其中一種,由Soumya Raychaudhuri實驗室開發。它是第一個系統性評估scRNA-seq整合方法的基準研究中最受矚目的方法之一。
Harmony的原理簡介:
Harmony使用模糊分群將每個細胞分配至多個群集中,對於每個群集,它計算各資料集需移動其該群集的質心至全域群集質心所需的修正量。由於每個細胞屬於多個群集,因此細胞的修正量是這些群集修正量的加權平均。這個過程會重複進行直到收斂或達到最大迭代次數。
- Harmony 在已經 PCA 降維過的空間上進行(例如前 20 個 principal components)。
- Harmony 將資料初始化為數個「暫時性群集」。這些群集依照 PCA 空間距離分配,並記錄每個細胞所屬的 batch ID(例如 sample A、B、C)
- 每一個迭代中:根據每個 cluster 裡不同 batch 的分布,計算 batch-specific centroid。將每個細胞的座標往一個「均衡位置」移動(weighted correction)
- 目標是:讓同一個 cluster 裡的細胞,不論來自哪個 batch,都更接近 cluster 中心
- 更新細胞對 cluster 的隸屬機率(soft assignment)
- 每個細胞可以同時屬於多個 cluster(機率形式),非硬性分群 → 重複這個過程數輪,直到收斂
- 最終輸出是 批次已修正的 PCA 向量(通常儲存在 harmony_embeddings)。 這個新空間可拿來做:
- RunUMAP() → 畫 UMAP
- FindNeighbors() → 建圖
- FindClusters() → 分群
Harmony提供一個可直接套用至Seurat物件的函數:RunHarmony。它接受先前合併的Seurat物件,並須指定哪一欄metadata作為批次來源:
library(harmony)
seurat <- RunHarmony(seurat, group.by.vars = "orig.ident", dims.use = 1:20, max.iter.harmony = 50)
seurat <- RunUMAP(seurat, reduction = "harmony", dims = 1:20)
seurat <- FindNeighbors(seurat, reduction = "harmony", dims = 1:20) %>%
FindClusters(resolution = 0.6)
saveRDS(seurat, file="integrated_harmony.rds")
dims.use設定要使用的PCA維度。
max.iter.harmony設定最大迭代次數。
plot1 <- UMAPPlot(seurat, group.by="orig.ident")
plot2 <- UMAPPlot(seurat, label = T)
plot3 <- FeaturePlot(seurat, c("FOXG1","EMX1","DLX2","LHX9"), ncol=2, pt.size=0.1)
((plot1 / plot2) | plot3) + plot_layout(width = c(1,2))
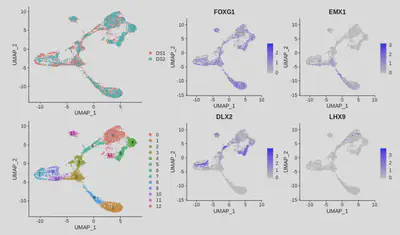
整合效果不錯,兩樣本的細胞彼此良好混合,並能觀察到一些發育軌跡。不過,對某些混合的群體(特別是非dorsal telencephalic細胞)仍需進一步判斷它們是否確為相同細胞類型。
由於Harmony以PCA結果作為輸入,並於PCA空間內進行修正,因此影響PCA的參數(如高變基因選取的nfeatures)亦會影響整合結果。RunHarmony預設會使用所有計算出來的主成分,但可透過設定dims.use加以指定。
需要注意的是,和CCA類似,如果細胞具有「生物上相似的表現特徵」,無論來自哪個 sample,它們會被聚在一起,弭平batch effect。但如果兩個samples是來自不同實驗條件,這樣的整合反而會干擾真正的生物變異,需要特別注意。
步驟2-3:使用LIGER進行資料整合
LIGER(Linked Inference of Genomic Experimental Relationships)是一種基於非負矩陣分解(Non-negative Matrix Factorization, NMF)的整合方法。它將每個資料集分解為共同與特有的因子矩陣 (NMF精髓),藉此找出跨資料集的共享細胞結構,同時保留每個資料集的獨特性。
LIGER整合步驟概要:
正規化資料集(通常以log-normalization處理)
將資料轉換為LIGER所需的格式(通常為liger::createLiger函數)
執行整合,包括optimizeALS()與quantile_norm()兩個關鍵步驟
使用整合後的空間做可視化與分群
library(liger)
# 將Seurat物件轉為list格式並取出表達矩陣
counts1 <- GetAssayData(seurat_DS1, slot = "counts")
counts2 <- GetAssayData(seurat_DS2, slot = "counts")
# 建立LIGER物件
liger_obj <- createLiger(list(DS1 = counts1, DS2 = counts2))
liger_obj <- normalize(liger_obj)
liger_obj <- selectGenes(liger_obj)
liger_obj <- scaleNotCenter(liger_obj)
liger_obj <- optimizeALS(liger_obj, k = 20)
liger_obj <- quantile_norm(liger_obj)
# 可視化與群集分析
liger_obj <- runUMAP(liger_obj, distance = 'cosine')
liger_obj <- findClusters(liger_obj, resolution = 0.4)
plotByDatasetAndCluster(liger_obj)
LIGER 偏好
使用原始counts資料,並自行處理正規化與篩選步驟。k為潛在因子的數目,是可調參數,應根據資料集大小與異質性設定。
步驟2-4:使用MNN(Mutual Nearest Neighbors)進行整合
MNN整合方法由Haghverdi等人提出,核心概念是尋找在不同資料集中互為最近鄰的細胞對,藉此計算校正向量並調整批次間的偏移。此方法由batchelor與scran套件實作。
MNN整合步驟:
將Seurat物件轉為SingleCellExperiment(SCE)格式
使用fastMNN()計算整合表達空間
將結果用於後續降維與分群
library(Seurat)
library(batchelor)
library(SingleCellExperiment)
library(scater)
# 轉換為SCE格式
sce1 <- as.SingleCellExperiment(seurat_DS1)
sce2 <- as.SingleCellExperiment(seurat_DS2)
# 使用MNN整合
sce.mnn <- fastMNN(sce1, sce2, d = 50, k = 20)
# 可視化與分群
sce.mnn <- runUMAP(sce.mnn, dimred = "corrected")
plotUMAP(sce.mnn, colour_by = "batch")
MNN需要先對每個資料集進行正規化(通常建議log-normalization)
d為PCA維度數,k為尋找MNN時的鄰近細胞數目
整合後的降維結果儲存在corrected中
小結與建議
本課程至此介紹了四種主流的scRNA-seq資料整合方法,包括:
Seurat(使用CCA與Anchoring機制)
Harmony(基於PCA的模糊分群與向量校正)
LIGER(基於NMF的共享與專屬因子分析)
MNN(尋找跨批次互為最近鄰的細胞進行修正)
這些方法各有優缺點,適用於不同情境,實作時建議:
若以Seurat為主流程,Seurat與Harmony較為便利整合
LIGER較適合於細胞異質性高或跨物種分析
MNN理論簡單,適用於數量不多但變異大的樣本
下一步,我們將示範如何比較整合後的結果、檢查整合品質(如kBET、LISI等評估指標),並進行下游分析(如差異表達、軌跡分析等)。
步驟3:整合品質評估與比較
整合的最終目的是讓相同細胞類型在不同樣本之間能夠準確對齊,同時保留真實的生物變異。因此,我們需要使用客觀指標來評估整合結果的品質。以下介紹幾個常見且被廣泛使用的指標:
3.1 kBET(k-nearest neighbor Batch Effect Test)
kBET 是一種基於最近鄰的統計測試方法,用來檢測在局部區域內不同批次細胞是否混合良好。理想情況下,不同批次細胞應在嵌入空間中彼此混合,若存在顯著的偏離,則說明仍存在批次效應。
可使用 kBET R 套件或 scIB(Python)來進行測試。
# 使用kBET評估
library(kBET)
kbet_result <- kBET(data_matrix, batch_labels, k = 30)
summary(kbet_result$stats)
指標:Rejection rate 越低表示混合越好(接近 0 為佳)。
3.2 LISI(Local Inverse Simpson’s Index)
LISI 衡量的是局部鄰域的多樣性,分為:
iLISI(Integration LISI):評估批次混合程度
cLISI(Clustering LISI):評估細胞類型區分能力
可使用 lisi 套件:
library(lisi)
lisi_scores <- compute_lisi(embedding, metadata, c("batch", "cell_type"))
iLISI 越高越好(表示批次混合良好)
cLISI 越低越好(表示細胞類型分離明確)
3.3 Silhouette Score
Silhouette Score 衡量細胞與其同群細胞間的距離是否小於與其他群細胞的距離。常用於評估群集清晰度。
library(cluster)
sil_scores <- silhouette(cluster_labels, dist(embedding))
summary(sil_scores)
分數範圍 -1 到 1,越高表示群集越分明。
步驟4:下游分析與應用
整合完成後,即可進行多樣的下游分析。以下列出幾個常見應用:
4.1 群集標記基因鑑定(Cluster Marker Identification)
DefaultAssay(seurat) <- "RNA"
markers <- FindAllMarkers(seurat, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
head(markers)
4.2 差異表達分析(Differential Expression Analysis)
可用於比較不同條件或群集之間的基因表現:
de_genes <- FindMarkers(seurat, ident.1 = "Cluster1", ident.2 = "Cluster2")
head(de_genes)
4.3 發育軌跡分析(Trajectory / Pseudotime Analysis)
可使用 Monocle3、Slingshot 等工具建立細胞狀態轉變軌跡:
library(monocle3)
cds <- as.cell_data_set(seurat)
cds <- cluster_cells(cds)
cds <- learn_graph(cds)
cds <- order_cells(cds)
plot_cells(cds, color_cells_by = "pseudotime")
課程小結
本課程從scRNA-seq整合的背景與必要性出發,依序介紹了四種整合工具(Seurat, Harmony, LIGER, MNN),並補充如何進行整合品質評估(kBET, LISI, Silhouette Score)以及典型的下游應用分析。透過這些工具與方法,研究者可以更準確解讀跨樣本、多條件的單細胞資料,為後續生物學發現與機制探討奠定基礎。