單細胞RNA-seq數據分析教學-2:細胞分群與初步標注
Quick look
在這堂課中,我們從FeaturePlot 初探基因表現,進一步進入圖論為基礎的細胞聚類,並完成初步的細胞型態標註流程。整體流程依序為:
- 使用 FeaturePlot() 觀察特定 marker 基因(如 CXCL9/CXCL10)在 UMAP 上的表現分布
- 基於圖的鄰接關係(KNN + SNN)建立細胞間的 network
- 利用 Louvain 或 Leiden algorithm 對細胞進行 community clustering
- 視覺化 clustering 結果並進行初步細胞標註
Feature plot
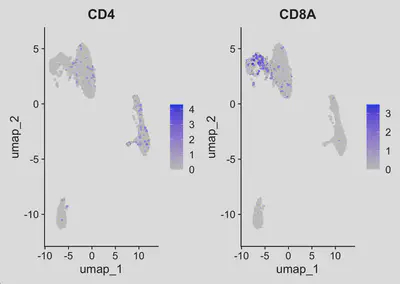
在做完數據降維及視覺化後,我們可以初步使用feature plots 來看某些目標基因的表現,讓我們能夠了解數據分佈的概況。
FeaturePlot(seurat, c('CXCL10', 'CXCL9'), ncol=2, reduction='umap')
Cluster the cells
雖然使用feature map 是一個不錯的開始,但是如果我們要更深入了解細胞的異質性,我們必須使用不具偏差的方式來標注細胞群。而clustering 就是第一步。原則上,我們可以任意選擇想要用的clustering algorithm,例如hierarchical clustering 與k-means,但實際運用比較困難,主要原因是scRNA-seq 數據通常較大,所以這些方法在效率上會比較慢。
除此之外,因為數據較為稀疏的關係,即便已經利用降維技術如PCA來降噪,細胞彼此之間的差異並不像bulk-RNA seq的數據可以比較好的量化。因此針對單細胞數據比較常用的方法為基於圖的聚類方法如graph-based community identification algorithm。
在這邊,圖(graph)是一個數學概念,基於一系列的物件、有關聯性的配對或是網路來進行擴展,而對單細胞數據而言就是細胞之間的網路。
至於為何要用圖論來解析高維空間的數據,原因如下:
scRNA-seq的背景雜訊(Noise)比例高 單細胞的樣本量小,導致每一個基因的表達值在技術與
生物變異上都會產生非常大的波動。 這使得即使已經做了PCA或其他降噪方法,細胞間的表達差異仍然難以明確區分。無法直接使用常見的距離衡量法 在bulk RNA-seq中,由於
表達值穩定,可以直接使用歐氏距離(Euclidean distance)或皮爾森相關係數等方式來比較樣本之間的相似性。
但在scRNA-seq中,因為資料稀疏與非線性,這些傳統的距離度量方法不再適用,導致需要依賴「圖論方法」如KNN graph + Louvain 或 Leiden algorithm來做聚類。Louvain 或 Leiden 這些方法會根據原始高維空間,或是指定的主成份數量中細胞間的 KNN 圖來做分群。它們是建構在保持局部結構的鄰接圖之上,不受降維影響。
第一步,先建立細胞間k-nearest neighbor network。每個細胞根據PC 數值先在空間中與最相鄰的細胞連接,接著會計算每一個細胞對(pair)之共同鄰居比例,來反應兩顆細胞之間連結的強度。強度不大的連結會被修剪掉,最後產生一個Shared Nearest Neighbor network。雖然看起來步驟有點複雜,但其實在Seurat中只有一行程式碼:
seurat <- FindNeighbors(seurat, dims= 1:20)
上面會建立一個網路,接者需使用louvain community identification algorithm 來在這個網路中尋找communities,也就是細胞因數據相似而靠在一起的群集。同樣Seurat也只提供一行程式碼解決:
seurat <-FindClusters(seurat, resolution=1)
這邊的resolution是用來控制要反回major cell types 還是更細微的cell subtypes。常用的值為0.1-1,通常可以微調來看哪一個數值比較符合分析的目的。數字越大解析度越好。
這邊需要注意的是,每做一次clustering 都會在seurat@meta.data 內出現新的分群資訊,如:
- RNA_snn_res.1
- RNA_snn_res.0.1 但seurat_clusters 與seurat@active.ident會是最新跑完的分群結果。
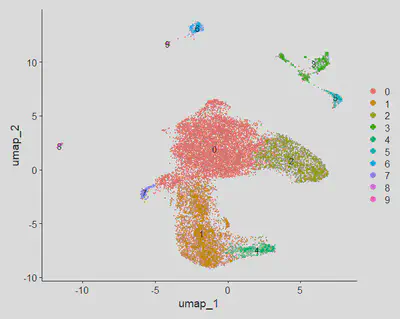
接著就是將分群結果視覺化了:
DimPlot(seurat, reduction='umap', label=TRUE)
細胞標注 (Cell annotaiton)
聚類後會給每一群細胞一個數字標記,所以我們可以假設同一個標注的細胞群彼此之間很相似,下一步就是要問某一群細胞到底是哪一種細胞?針對這個問題目前沒有一個完美的答案,輔助標注的工具眾多且各有優缺點,但有幾個原則可以先注意:
- 先檢查canonical cell type的基因表現。
- 找出signature genes 或是marker genes,可以靠尋找文獻、enrichment analysis或是詢問專家。
- 和現行的參考數據做比較。
手動標記基因檢查
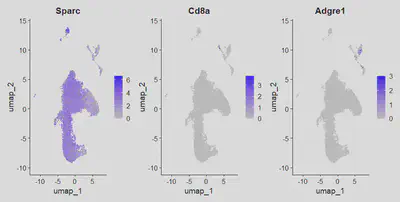
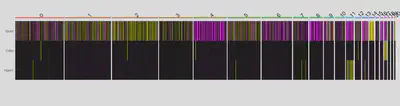
我們這邊使用mouse攝護腺癌細胞當作範例,單純看CD8+ T 細胞、巨噬細胞與fibroblast marker (因為RM1 prostate cancer cells 具有fibroblast-like morphology,所以用fibroblast marker 可以迅速找到,但還是需要後續分析實際marker,這邊只做舉例)。最後可以用Heatmap 來觀察這些基因表現在每一群細胞的狀況。
# 定義標記基因列表
markers <- c("CD8a", "Adgre1", "Sparc")
FeaturePlot(seurat, features = markers, ncol=3, reduction='umap')
DoHeatmap(seurat, features=markers) + NoLegend()
接下來,為了要讓細胞標注不受到任何偏差影響,我們必需要針對每一個細胞群找出cluster markers。在Seurat,只要使用FindAllMarkers()函式即可:
cl_markers <-FindAllMarkers(seurat, only.pos=TRUE, min.pct=0.25, logfc.threshold=log(1.2))
library(dplyr)
cl_markers %>% group_by(cluster) %>% top_n(n=5, wt=avg_logFC)
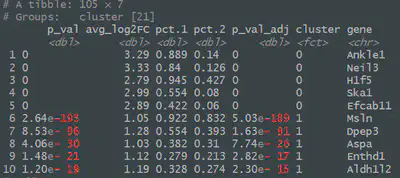
因為每一顆細胞都代表一個樣本,所以建議也要調整在群集內與外的基因的偵測率(pct)與fold change between cells (logfc)。
上面的過程會花不少時間,但我們可以使用presto這個套件來加速分析:
# 安裝
remotes::install_github("immunogenomics/presto")
# 使用
markers <- wilcoxauc(seurat, group_by = "seurat_clusters")
top_markers <- markers %>% group_by(group) %>% top_n(5, auc)
我們可以發現結果是很類似的,但速度更快。
課程小結
Feature Plot 初探 利用 FeaturePlot() 可視化基因在 UMAP 上的表現,有助於初步了解細胞間的異質性及 marker distribution。
為什麼要用圖論來聚類?
- scRNA-seq 資料稀疏且雜訊高,常見的距離計算方法難以準確反映細胞間差異
- 圖論(KNN graph)能保留局部結構,有助於辨識生物學上相似的細胞群
- 用 Seurat 建構細胞之間的 SNN network 並執行 Louvain 聚類
- FindNeighbors() → 建立 shared nearest neighbor network
- FindClusters() → 執行 Louvain clustering,可調整 resolution 解析度
- 細胞標註(Cell Annotation)策略
- 利用 canonical markers(如 CD8a、Adgre1、Sparc)進行手動標註
- FindAllMarkers() 可快速篩選各 cluster 的差異性基因
- 搭配 presto 套件(使用 AUC-based ranking)可加速大資料量運算,效果與傳統 Wilcoxon 方法相近但更高效
- 視覺化推薦:
- 使用 DoHeatmap() 或 FeaturePlot() 快速查看 marker gene 在群集中的分布情況
- 適當設定 min.pct、logfc.threshold 以控制 marker gene 的可靠性