單細胞RNA-seq數據分析教學-1:從數據處理到降維視覺化
Quick look
這篇參考Zhisong He與Barbara Treutlein等人撰寫的單細胞定序處理教學。
安裝與導入必要套件
# 基礎分析套件
install.packages("Seurat")
install.packages("dplyr")
install.packages("patchwork")
install.packages("ggplot2")
# 輔助套件(用於快速標記基因分析)
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("presto") # 快速Wilcoxon檢定
# 加載套件
library(Seurat)
library(dplyr)
library(patchwork)
library(presto)
創建Seurat物件
從10x Genomics數據讀取並構建Seurat物件:
# 讀取10x數據
counts <- Read10X(data.dir = "scRNA_data/scRNA_project1")
seurat <- CreateSeuratObject(counts, project = "scRNA_project1")
# 手動讀取非10x數據(例如矩陣文件)
counts <- readMM("scRNA_data/scRNA_project1/matrix.mtx.gz")
features <- read.csv("scRNA_data/scRNA_project1/features.tsv.gz", sep = "\t", header = FALSE)
colnames(counts) <- readLines("scRNA_data/scRNA_project1/barcodes.tsv.gz")
rownames(counts) <- make.unique(features[,2])
seurat <- CreateSeuratObject(counts, project = "scRNA_project1")
關於讀取要使用的函式,如果資料夾裡面是以下文件,使用Read10X()
matrix.mtx.gz(稀疏矩陣格式的基因表達數據)features.tsv.gz(基因名稱與編號)barcodes.tsv.gz(細胞條碼資訊) 通常只有一行,所以很適合用readLines來逐列讀取,存成character向量。 這些檔案通常是 gzip 壓縮格式(.gz),但 Read10X() 也可以讀取未壓縮的版本(即 .mtx、.tsv)。
如果資料夾裡的檔案是CellRanger 處理好的輸出: filtered_feature_bc_matrix.h5,使用Read10X_h5()
品質控制(QC)
這一步我們希望過濾掉:
- 偵測到太少基因的細胞:常見原因是測序深度太淺,通常研究需要2-2.5倍的測序深度 (1倍為10000 reads per cell)。
- 偵測到太多基因的細胞:可能原因為一顆油滴包到兩顆以上的細胞 (doublet or multiplet),所以共享cell barcode,導致基因數量大增。
- 太多粒線體的轉錄本:大多的scRNA-seq實驗是用oligo-T去抓mRNA,理論上不會抓到粒線體的基因,因為粒線體轉錄本大多缺少穩定的poly-A tail(較短且角色較複雜),但難免會抓到一些。也有證據顯示一些粒線體的轉錄本帶有poly-A tails作為降解的標記。原則上,太多粒線體基因表示細胞處於stress (如缺氧),所以產生更多粒線體。
而Seurat可以基於以下指標,過濾低質量細胞:
nFeature_RNA:每個細胞檢測到的基因數nCount_RNA:總轉錄本數percent.mt:粒線體基因比例
以下數據我用我之前在國衛院上課時給的pbmc數據做範例。
針對粒線體基因比例需要手動設定:
# 計算線粒體基因比例
seurat[["percent.mt"]] <- PercentageFeatureSet(seurat, pattern = "^MT-")
請注意,針對需要過濾的基因數量,並不存在one-size fits all的過濾條件,所以建議過濾掉outlier cells 即可,也就是明顯不屬於大部分細胞的細胞群。因此我們可以先視覺化基因分佈圖:
# 可視化QC指標
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
# 過濾細胞(根據分佈調整閾值)
seurat <- subset(seurat, subset =
nFeature_RNA > 500 &
nFeature_RNA < 5000 &
percent.mt < 5
)
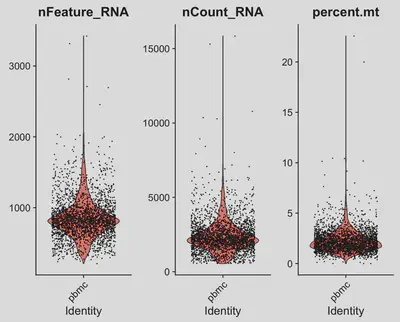
請記住,上面的數值要根據跑出來的Violin plots來決定。 另外,有時候也建議做額外的QC,尤其是針對doublets的偵測。主要原因是,因為不同的細胞抓到的RNA變化很大,所以即使邏輯上doublets 的基因會比單顆細胞高出很多,也不能保證能藉此排除doublets的可能! 目前有一些工具來預測某細胞究竟是singlet 或是doublet。例如DoubletFinder,利用隨機平均化細胞來建立人工doublets,然後針對每顆細胞去測試與人工doublets的相似程度。除此之外,粒線體的轉錄本比例可能並不足夠完全排除不健康或是受到壓力的細胞,有時候就會需要其他方式來過濾,例如使用機器學習的方式來做預測。
數據標準化
與bulk RNA-seq一樣,每顆細胞的基因轉錄本數量都不同,也就是被抓到的RNA數量不同,因此不能直接比較。舉個例子,假設今天有兩位攝影師在拍鳥類生態紀錄片,一位待在鳥類豐富的森林,一位則在鳥相較稀少的郊區。他們分別拍攝了 300 張與 50 張鳥類照片。乍看之下,拍到 300 張的攝影師似乎捕捉到了更多鳥類,但如果我們考量他們待在現場的時間——前者拍攝了 10 小時,而後者僅拍攝了 2 小時,那麼計算每小時拍攝的鳥類照片數量,分別是 30 張/小時與 25 張/小時。這樣的標準化比較才更具代表性,而不是僅看總照片數。因此,在分析單細胞 RNA-seq 數據時,通常需要進行歸一化,以確保比較不同細胞間的基因表達量時,不受總 RNA 捕獲量的影響。
針對scRNA-seq,非常常用的標準化方法與TPM (Transcripts per million reads)的方式很類似,也就是針對每顆細胞的總表現亮做歸一化,然後再乘上一個縮放因子 (default 為10000),公式如下:
其中:
- Xij是細胞i中基因j的原始 UMI(Unique Molecular Identifier)計數。
- 分母是是該細胞 所有基因的 UMI 總和,也就是該細胞的測序深度。
- 取log目的是將基因表現分佈拉到常態分佈。
- 乘上
10000用來將比例數據擴展到更容易比較的範圍(類似於 TPM)。 - 加 1(又稱
pseudocount) 避免 log 取值為負無窮大,或是出現zero transcripts。
# 標準化(LogNormalize)
seurat <- NormalizeData(seurat)
通常NormalizeData()有一些設定參數,但一般不用去更動,default 即可滿足大部分分析的需求。
特徵選擇
並非細胞裡面的所有基因都有相同程度的訊息,例如低度表現得基因,或是不同細胞間表現程度相近的基因,基本上並不帶有可用的訊息,因此需要利用特徵選取來找出具有代表性的基因。不過別擔心,Seurat的FindVariableFeatures()可以幫我們找出具有高度變異性的基因。
# 識別高度可變基因(HVGs)
seurat <- FindVariableFeatures(seurat, nfeatures = 3000) #可以2000-3000視情況而定
# 可視化HVGs
top20 <- head(VariableFeatures(seurat), 20)
plot <- VariableFeaturePlot(seurat)
LabelPoints(plot, points = top20, repel = TRUE)
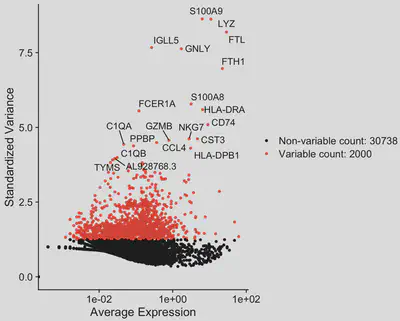
Seurat 針對不同樣本的某基因,計算標準化的變異 (standardized variance),並選取前2000-3000個基因。
值得注意的是,關於要選多少基因,目前並沒有好的準則,有時候需要選擇不同的數量來看分析效果,並選擇最能解釋結果的基因數量。大致上,2000-5000個基因是可以接受的範圍,選擇不同的基因數量並不太會影響分析結果。但根據最新的論文,2000個基因是最好的選擇。
數據縮放
因為不同的基因有不同的base expression 與分佈,因此每個基因的貢獻程度是不同的。為了不讓某些基因的影響力偏移,我們需要做適當的縮放。
# 縮放數據(可選回歸變異源)
seurat <- ScaleData(seurat, vars.to.regress = c("nFeature_RNA", "percent.mt"))
SCTransform
上面展示的一般的正規化與縮放流程,利用NormalizeData()與ScaleData()來調整基因數據到可以比較的形式,然而傳統的log-normalization轉換會有個缺點,就是會導致零膨脹效應(zero-inflation artifact)。這個現象是在說明當數據裡面出現過多的零時 (可預期或是不可預期),會影響統計模型的建立與後續的預測,因此有不少的演算法在解決這一塊。而對於單細胞數據來說,基因的dropout本來就是常見的現象(以10X來說),因此拿到的數據會有大量的零,而這些零在log-normalization後雖然仍為零(log(x+1)),但是會被拉近與其他基因表現的距離,因此會導致零這個數值影響力變大。為了避免這個問題,Hafemeister與Satija教授開發了SCTransform,利用一regularized negative binomial regression model 來fit基因的表現,同時考慮測序深度(將不同基因的UMI做考慮,不像TPM那樣弭平測序深度的影響)。
技術上:
- 基於 Pearson 殘差(Pearson residuals)變異性來選擇高變異性基因。
- 自動對測序深度進行正規化(即不需要手動調整 UMI 數)。
- 使用一般化線性模型(GLM, Generalized Linear Model)調整技術變異。
seurat <- SCTransform(seurat, variable.features.n = 2000)
#也可以選擇要將哪些特徵作regress out,近一步移除不想要的數據變異
seurat <- SCTransform(seurat,
vars.to.regress = c("nFeature_RNA", "percent.mt"),
variable.features.n = 2000)
這個函式基本上已經包含了normalization、scaling與特徵選取的過程,所以要注意若使用SCTransform,不需要做上面的步驟。然而SCTransform有一些缺點:
- 效率相對慢。
- 傳統log-normalization 的正規化是針對dataset裡每個細胞做計算,正規化後數據可以不依賴測序深度,因此不同datasets之間的細胞是可以互相比較的,即使沒有辦法處理
測序深度的影響以及校正生物或是技術噪音。但SCTransform 需要考慮dataset裡其他細胞的資訊來做計算,然而不同datasets的細胞資訊都不同,所以兩個各自使用SCTransform處理的數據是無法互相比較的,如果整合後需要做differential expression analysis需要另外處理。我們會在數據整合(integration)的時候專篇講解。
線性降維
雖然我們可以在找到高變異性基因後觀察細胞異質性,但強烈建議在標準化之後先使用線性降維來對數據做整理。這個步驟在目前單細胞分析算是基本需求,因為:
- 數據變得較為
緊密(compact),運算更快。 - 因為單細胞數據內在較為稀疏,在降維處理後可以增強特徵訊號的穩健性。
而PC的數量取決於基因或是細胞的數量,看哪一個數量比較少。然而大多數PC並不會提供太多訊息而只是隨機噪音,只有前幾個PC帶有較多的訊息來解釋細胞之間的特徵變化。Seurat 在計算PCA時並不會計算全部的PC,而是只針對前50 PC 做運算(by deafult),但這個數字可以透過
npcs參數去做修改。
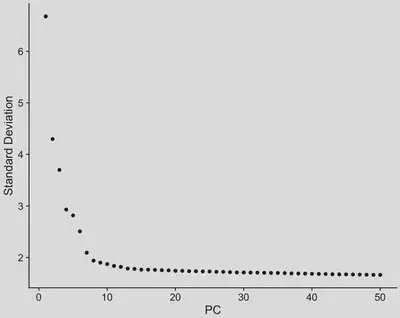
但到底要選擇多少PC才夠?
# PCA降維
seurat <- RunPCA(seurat, npcs = 50)
# 可視化PCA結果(肘部圖)
ElbowPlot(seurat, ndims = 50)
# 選擇前20個主成分進行後續分析
PCHeatmap(seurat, dims=1:20, cells=500, balanced=TRUE, ncol=4)

非線性降維與分群
scRNA-seq分析通常基於多個基因的表達值來比較細胞。例如,聚類分析 (clustering) 透過計算細胞在多個基因上的歐幾里得距離,來識別具有相似轉錄組特徵的細胞。在這些應用中,每個基因都可以視為一個數據維度。舉個簡單的例子,假設我們的數據集中僅包含兩個基因,那麼我們可以用二維圖來表示,每個軸對應一個基因的表達量,而圖上的每個點代表一個細胞。當數據集包含數千個基因時,每個細胞的表達譜就構成了一個高維表達空間中的位置。
降維 (dimensionality reduction) 的目標是減少數據的維度數量,以便進行更高效的分析。
這是因為許多基因的表達是相關的,受到相同的生物過程影響,因此無需單獨存儲每個基因的資訊,而是可以將多個基因的特徵壓縮到少數幾個統合的維度,例如特徵基因 (eigengene)(Langfelder 和 Horvath,2007)。降維的主要優勢包括:
- 減少計算負擔,使後續如聚類等分析僅需在少數維度上運算,而非上千個基因。
- 降低數據噪聲,透過整合多個基因的資訊來獲得更穩定的模式表現。
- 便於數據可視化,讓我們能夠在 2D 或 3D 圖 中直觀展示細胞間的關係,避免高維度的數據難以理解的問題。
而且降維後,會將稀疏矩陣轉換成較緊密的數據(compact),有助於後續的分析的效率與可靠性。這種方式對分析基本上沒有缺點,除非是整合百萬細胞數量等級的數據,可能需要保存稀疏矩陣的特性來減少記憶體的佔用 (可以用truncated SVD 或是 autoencoder)。
透過降維技術,如主成分分析 (PCA) 和 t-SNE、UMAP 等非線性方法,研究者可以更有效地探索單細胞數據的內在結構,發現潛在的細胞類型與功能模式。
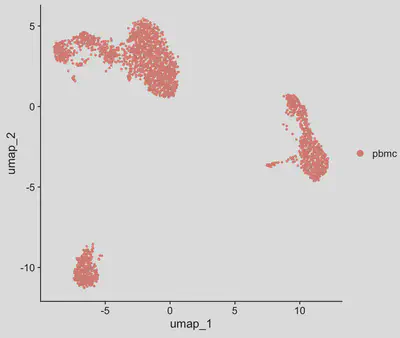
# UMAP降維
seurat <- RunUMAP(seurat, dims = 1:20) # UMAP 資訊會自動存在seurat裡更新
UMAPPlot(seurat)
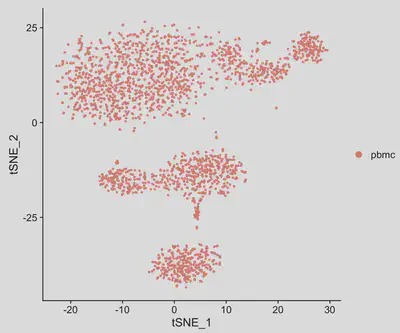
# TSNE降維
seurat <- RunTSNE(seurat, dims = 1:20) # UMAP 資訊會自動存在seurat裡更新
TSNEPlot(seurat)
結論
本篇教學文章介紹了單細胞RNA定序(scRNA-seq)分析的基礎流程,從數據讀取、Seurat物件建立、品質控制、標準化與特徵選取,到降維與視覺化,構成了典型的單細胞分析前處理與探索性分析流程。透過這些步驟,我們能有效地過濾低品質細胞、識別具有生物學意義的基因表達變化,並在降維空間中觀察細胞間的異質性與潛在群集結構。
值得強調的是,scRNA-seq數據分析並非僅依賴單一工具或固定參數,每個步驟中都蘊含著根據實驗設計、樣本特性與研究問題進行調整的彈性與必要。例如,品質控制的閾值應根據實際資料分佈決定;HVGs的選取數量與方法則可能影響下游分析結果。此外,選擇何種正規化方式(如LogNormalize或SCTransform),也需根據是否需要後續整合多個樣本或資料集而做考量。
後續的教學將會進一步介紹:細胞分群(clustering)、細胞類型註解(cell type annotation)、差異表達分析(differential expression)、軌跡推斷(pseudotime trajectory)、資料整合(integration)以及細胞通訊(cell-cell communication)等更進階的主題。
單細胞分析不僅為基礎研究提供了前所未有的解析度,也為轉譯醫學與個人化醫療開創了全新視野。希望透過這一系列教學,能幫助讀者打下堅實的分析基礎,並在未來更有效地應用這項技術於自己的研究之中。