迷你課程:R語言生物資訊實作~Bulk RNA-seq分析-1

Quick look
歡迎來到我們的bulk RNA-seq分析課程,在這裡我們將把原始的基因表達計數數據轉化為有意義的生物學發現。
為什麼Bulk RNA-seq分析如此重要
RNA測序技術(RNA sequencing, RNA-seq)透過對全轉錄組的高通量分析,為我們提供了在不同生理條件、實驗處理及疾病狀態下基因表達的全景圖,從而徹底革新了我們對細胞功能與基因調控的理解。相較於傳統只能針對少數基因進行分析的方法,RNA-seq能夠同時量化約20,000個人類基因的表達水平,使研究人員能夠以系統性方式解析細胞內的轉錄活動,發掘潛在的生物標記與調控機制。
生物學影響
- 疾病研究:RNA-seq揭示了在癌症、神經系統疾病、自身免疫性疾病等無數其他疾病中哪些基因失調。搭配生物資訊學的其他工具如路徑分析,這些發現直接導致新的治療靶點和診斷標記的發現。
- 藥物開發:當製藥公司測試新療法時,RNA-seq顯示藥物如何影響細胞訊息傳遞路徑,幫助優化劑量、識別恰當的生物標記物並預測副作用。
- 精準醫學:通過了解個體患者的基因表達譜如何對不同治療產生反應,臨床醫生可以藉由個人化的治療模式以獲得更好的結果。
- 基礎生物學:RNA-seq可以幫助闡述了發育、衰老、應激反應和細胞分化等基本過程,擴展了我們對生命在分子階層上如何運作的理解。
分析挑戰
雖然RNA-seq技術能夠產生大量涵蓋整個轉錄組的有用數據,但從中提取具生物意義的訊息並不直接,往往需要結合複雜的統計方法與計算模型。這是因為基因表達數據本身具有以下幾個重要特徵:
高維度(High Dimensionality): RNA-seq可同時量化數千至數萬個基因的表達水準,遠多於樣本數。這種「大p小n」的特性(即變數遠多於樣本)對統計分析構成挑戰,容易導致過擬合或偽相關,需要降維、正則化等方法來提高模型的穩定性與可解釋性。噪音大(High Noise Levels)基因表達受到技術層面(如擴增效率、批次效應)及生物學層面(如細胞異質性、時序變化)等多重變因影響,導致測得的表達數據中包含大量背景噪音,可能掩蓋真實的差異訊號。因此,需要進行嚴謹的資料預處理,包括去除低表達基因、批次校正與正規化等步驟,以提取出可靠的生物學資訊。複雜依賴性(Complex Dependencies)基因並非獨立運作,而是在調控網絡中以協同、互作、反饋等方式影響彼此。例如,轉錄因子調控目標基因表達,或多基因參與相同生物路徑。這種結構性使得簡單的統計假設(如獨立性)不再適用,分析需考慮基因間關聯,例如利用共表達網絡、基因集富集、分析(GSEA)或圖論方法。上下文依賴性(Context Dependence)基因表達模式高度依賴於細胞類型、發育階段、生理狀態及外界刺激等條件。在一個特定的組織中活躍的基因可能在另一個環境下完全沉默。因此,在分析RNA-seq資料時,必須將樣本的生物學背景納入考量,否則容易導致錯誤推論。例如,同一基因在正常組織和腫瘤中的表達差異,往往反映出疾病特異性的轉錄調控變化。
差異表達分析(DEG Analysis):現今主流方法概覽
在 RNA-seq 資料分析中,差異表達分析(Differential Expression Analysis, DEG)是最核心的任務之一。它幫助我們找出在不同實驗條件(例如疾病 vs 健康、處理 vs 控制)下,表達量有顯著改變的基因。為了應對 RNA-seq 數據的特性(如離散性、高變異與多重檢定),研究者已發展出多種統計方法。以下為目前最常用的幾種:
| 方法 | 核心模型 | 適用對象 | 優點 | 缺點 |
|---|---|---|---|---|
| DESeq2 | 負二項分布 + shrinkage | Bulk RNA-seq | 保守穩健、適合小樣本、廣泛使用 | 計算稍慢、不適用單細胞資料 |
| edgeR | 負二項分布 + 精確檢定 | Bulk RNA-seq | 快速、靈活、支援複雜設計 | 對低表達基因較敏感 |
| limma-voom | 線性模型 + voom 轉換 | Bulk RNA-seq(類似微陣列分析) | 適合大樣本與複雜設計 | 對低計數資料表現較差 |
| baySeq | 貝葉斯模型 | Bulk RNA-seq | 適用先驗知識 | 使用上相對複雜 |
| NOISeq | 非參數法 | Bulk RNA-seq | 不依賴分布假設 | 對樣本數需求高 |
| MAST, SCDE, Seurat (FindMarkers) | 混合模型、貝葉斯、Hurdle 模型等 | 單細胞 RNA-seq | 特化單細胞特性 | 不適用 bulk RNA-seq |
DESeq2 — Shrinkage (收縮估計) DESeq2 同樣使用 負二項分布,但強調在估計 dispersion 以及 fold change (log2FC) 時加入 Bayesian shrinkage。 它的理念是:小樣本下直接估計 fold change 容易不穩定,因此應向平均值「收縮」(借力於其他基因)。 這使用 empirical Bayes shrinkage estimation(尤其是 log2 fold change 的 shrinkage,如 lfcShrink() )。 優點:保守、穩健,特別是在少樣本下提供更穩定的 fold change 與 p 值 缺點:速度略慢;對於極端差異基因可能會「低估 fold change」
edgeR — 精確檢定 (Exact Test) edgeR 假設基因表現遵循 負二項分布 (Negative Binomial, NB),來建模 RNA-seq 計數的離散性與過度變異 (overdispersion)。 它使用 精確檢定 (exact test),這是一種類似 Fisher’s exact test 的方法,針對 小樣本下的兩組比較,在 NB 分 布下計算 p 值。 若設計更複雜(>2 組或加上 batch),會使用 generalized linear models (GLM)。 優點:非常適合小樣本快速進行簡單兩組比較 缺點:小樣本時對於 dispersion 的估計容易不穩,需要良好的 library size normalization。
為什麼 DESeq2 成為經典標準?
想像你正比較健康與疾病組織樣本的基因表達,從數千個基因中得到的 RNA 計數堆滿整張表格。問題來了:哪些基因的變化是真正的生物現象?哪些只是噪音? 這正是 DESeq2 大放異彩的地方。它不只是另一種分析工具,而是一個專為 RNA-seq 數據打造的統計框架。傳統統計方法假設資料連續、常態分布,但 RNA-seq 的世界混亂又嘈雜,基因表達高變異、低計數、技術誤差多,DESeq2 正是為這樣的現實而誕生。 經過十年、大量文獻與研究實踐的驗證,DESeq2 已成為最值得信賴的差異表達分析工具之一。
為什麼 RNA-seq 數據如此特別?
在理解 DESeq2 如何運作前,先來看看為什麼 RNA-seq 資料如此「難搞」,以致於不能用一般統計方法處理:
計數資料(Count Data)不是連續變數: RNA-seq 給我們的是每個基因的 RNA 分子數量,都是整數。例如你不會有 2.5 個 RNA,只會是 2 或 3。這讓很多傳統統計假設
(如常態分布)不再成立。基因之間表現量差異巨大 有些基因每個樣本會產生上千個 RNA 分子,而有些基因則只有幾個。舉例來說,一個基因平均表現量是 10,在不同樣本間可能落在 8 到 12 之間;但另一個基因如果平均有 1000,在樣本間可能會介於 800 到 1200。這不是錯誤,而是生物本來就會這樣運作!但這也代表:
不同基因的變異尺度差異很大,我們不能用同一種統計模型套用到所有基因上。定序深度的差異 即使樣本準備得很小心,有些樣本的定序結果還是會比其他樣本多出很多
reads。舉例來說,有的樣本可能產出 2 千萬個 reads,而有的樣本則有 3 千萬。這種差異是技術性差異,不是生物造成的,但卻會影響每一個基因的計數值。 所以我們必須進行樣本之間的歸一化(normalization),否則這種深度差異會被誤認為是基因表現變化。多重檢定的陷阱 RNA-seq 通常是同時比較 20,000 個以上的基因。如果我們對每個基因都用傳統的顯著性水準(例如 p 值 < 0.05),那在完全沒有任何差異的情況下,也會有大約 1,000 個基因「看起來」有差異,純粹是
隨機產生的假陽性。 這就是所謂的多重檢定問題(multiple testing problem),所以在分析時必須進行FDR(假發現率)校正,以避免誤判結果。
DESeq2 基本流程
第一步: Size factor 的評估
首先我們在測序完之後,必須要根據每個樣本的測序深度做檢驗,也就是要去評估size factor,DESeq2會針對計算出來的size factor對樣本的測序資料做歸一化:
Before normalization:
sample1 <- c(100, 200, 50) # 20M total reads
sample2 <- c(150, 300, 75) # 30M total reads
After size factor normalization:
sample1_norm <- c(100, 200, 50) # Reference
sample2_norm <- c(100, 200, 50) # Scaled down by factor of 1.5
第二步: Dispersion 的評估
RNA-seq 輸出的reads高達上百萬,而每個基因的counts變異很大,但都約為10-1000左右,因此對某一個read要能對應上某一個基因的機率是相對很低的。這種離散的分佈、低機率加上大量的對應 (reads mapped to a gene)其實就跟普瓦松的分佈非常類似,當在二項分佈中試驗的次數變多,而試驗成功的機率變低時,分佈就可以用普瓦松分佈來趨近。然而若要使用普瓦松分佈,就必須要符合平均值等於變異數的性質 (依照公式可得出此結論),但RNA-seq的數據並非如此,原因是不同基因的樣本間技術上生物變異大,而針對mean與variance作圖時會發現overdispersion的現象 (圖一),並不適用於普瓦松分佈?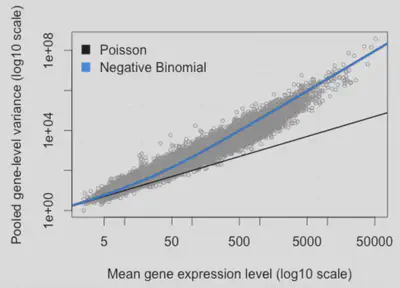
DESeq2 會估算某基因計數在樣本間的變異程度,這個步驟非常重要,因為針對變異較大的基因,需要更強的證據來說明他為differentially expressed,我們才成正確的找到具有生物意義的基因變化。
第三步: 統計模型
在完成上面步驟後,DESeq2就可以來用統計模型估算基因表達的統計差異了。
我們在正式進入分析前,先回顧一下名詞:
Size factor: 就是標準化的權重,如果有一樣本size factor 為1.5,就表示他比平均值在測序上深了50%。
Dispersion: 主要是用來描述樣本間基因的變異程度。變異程度越大的基因表示雜訊越多,變異程度小的基因表示樣本間表現平穩。
Log2 Fold Change: 量化基因表達改變的指標,也是DE分析的
effect size。
Adjusted P-value: 因為DE分析同時比較了近兩萬個基因,所以這在統計上是一種多重比較,需要用一些方式來控制偽陽性率,如果adjusted P-value 為0.05,表示目前此基因的偽陽性率是5%,所以這個數值越低越好。
準備好把 RNA-seq count 檔案轉換成 DESeq2 可以直接分析的物件了嗎?這就是從理論到實作的關鍵一步:把序列分析 pipeline 的輸出,整理成統計模型可以用的輸入。 一旦掌握核心要素,DESeq2 的分析其實非常直覺!
需要準備的兩個檔案
- Count Matrix(計數矩陣)
- 列(row):基因
- 欄(column):樣本
- 值(value):整數型計數(不能有小數!)
- Sample Metadata(樣本資訊表)
- 列(row):樣本(名稱需與 count matrix 的欄名對應)
- 欄(column):實驗條件(condition, batch, timepoint 等) 👉 就這兩樣!把它們準備好,就能開始組裝分析物件。
核心流程:三步驟
第一步:載入套件與資料
載入 DESeq2
library(DESeq2)
匯入 count matrix
#讀取count matrix,注意csv檔的第一欄就是基因名稱,所以在讀取的時候row.names=1,第二欄開始為sample name
#讀取進來是data frame的形式
counts <- read.csv("count_matrix.csv", row.names = 1, check.names = FALSE)
counts <- as.matrix(counts) #轉成矩陣,運算效率更快
storage.mode(counts) <- "integer" # 確保為整數,因為是`counts`
匯入樣本資訊
metadata <- read.csv("sample_info.csv", row.names = 1, stringsAsFactors = TRUE)
確認樣本名稱一致
stopifnot(identical(colnames(counts), rownames(metadata)))
第二步:建立 DESeqDataSet 物件
dds <- DESeqDataSetFromMatrix(
countData = counts,
colData = metadata,
design = ~ condition # 依照實驗設計調整
)
第二步:基本前處理
過濾低表達基因
keep <- rowSums(counts(dds)) >= 10 #會找出每一列,也就是每一個基因的counts總和需要≥10
dds <- dds[keep, ]
# 設定對照組為參考 (reference level)
dds$condition <- relevel(dds$condition, ref = "control")
完成!現在你已經擁有 分析-ready 的 DESeqDataSet 🎉
三大地雷要避免
- 樣本名稱不一致
identical(colnames(counts), rownames(metadata)) # 必須 TRUE
- 計數不是整數
all(counts == round(counts)) # 必須 TRUE
- 參考組設錯
levels(dds$condition) # 檢查因子水平
dds$condition <- relevel(dds$condition, ref = "control")
快速 QC
概覽資料
dds
每個樣本的測序深度
colSums(counts(dds))
每個基因至少在幾個樣本被偵測到
rowSums(counts(dds) > 0)
儲存以便後續分析
saveRDS(dds, "analysis_ready_dds.rds")
課程小結
你已完成的目標
- 了解 bulk RNA-seq 的價值與常見挑戰(高維度、雜訊、依賴性、情境依賴)。
- 掌握差異表達分析主流方法的版圖,聚焦 DESeq2 的核心思路(負二項 + shrinkage)。
- 準備好兩個關鍵輸入:Count Matrix 與 Sample Metadata,並建立 DESeqDataSet。
- 進行最小可行的 前處理(低表達過濾、參考組設定)與 基本 QC(深度、偵測度)。
本章關鍵觀念
- Size factor:校正測序深度差異,避免把技術性差異誤當生物訊號。
- Dispersion:度量跨樣本變異,DESeq2 以經驗貝葉斯估計穩定變異與效應量。
- Shrinkage:在小樣本下「收縮」log2FC,使估計更穩健、可重現。
- 多重檢定(FDR):同時檢驗上萬基因必須控制偽陽性。
實作檢核清單
- colnames(counts) == rownames(metadata)
- storage.mode(counts) == “integer”(全為整數)
- 低表達過濾門檻(如 rowSums(counts(dds)) >= 10)已應用
- 參考組以 relevel() 正確設定(如 ref = “control”)
- 物件已保存:saveRDS(dds, “analysis_ready_dds.rds”)
常見地雷
- 樣本對不上:欄名與列名任何一處空白/符號/大小寫不一致都會失敗。
- 非整數計數:TPM/FPKM 不能直接用於 DESeq2;請使用原始 counts。
- 設計式錯誤:忘記把批次或時間點寫入 design,將導致錯誤推論。
- 過度過濾或過度寬鬆:低表達基因過濾門檻過高會犧牲訊號,過低易放大雜訊。